The EU GMO Framework Is Slowing Down Gene Therapy. Here's What Regulatory Affairs Teams Need to Do About It.
You've secured your CTA. Your trial is designed. Your sites are ready.
Then the GMO clock starts — and suddenly you're looking at up to 12 months of additional review before a single patient can be treated.
If you work in regulatory affairs for gene therapy medicinal products (GTMPs), this is not a hypothetical. It's a recurring reality that continues to delay access to potentially curative therapies for patients with serious unmet medical needs.
Here's a clear-eyed breakdown of where the framework stands today — and what you can do to navigate it more effectively.
Why the EU GMO Framework Was Never Built for Medicine
The EU GMO regulatory framework — primarily Directive 2001/18/EC on deliberate release and Directive 2009/41/EC on contained use — was designed to protect agricultural ecosystems and food consumers. It was not designed with gene therapy clinical trials in mind.
Yet GTMPs that contain or consist of GMOs must comply with both. The result is a parallel approval pathway that sits entirely outside the Clinical Trials Regulation (CTR) 536/2014 — and one that varies dramatically between member states.
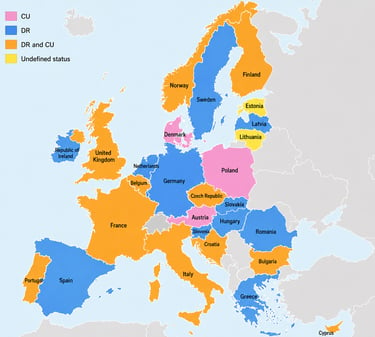
The classification matters enormously in practice:
• Contained use (CU) applies physical or combined barriers to limit GMO-environment contact. It has a structured risk classification (Class 1–4) that enables somewhat shorter review timelines.
• Deliberate release (DR) applies when no specific containment measures limit environmental contact — which includes considerations around virus shedding and replication. Regulatory procedures here are more stringent and often significantly longer.
Some products fall under both directives, adding another layer of complexity to dossier preparation and submission strategy.
The Framework Each EU Country Applies Is Not the Same
This is one of the most underappreciated sources of variability in EU gene therapy development: member states have not only transposed the EU directives differently into national law — many have actively chosen which framework applies to GTMPs on their territory.
Spain, for example, has adopted a DR-only framework for GTMP-GMO clinical trials. Sponsors submitting there do not need to navigate deliberate release requirements at all.
Italy, by contrast, operates under both frameworks depending on the product and trial design — contained use for genetically modified microorganisms under Directive 2009/41/EC, and deliberate release provisions where applicable under 2001/18/EC.
This means the technical file your team prepares — and the ERA it must support — differs not just in timeline but in substance between countries. The dossier requirements under Directive 2001/18/EC (deliberate release) are more extensive: they include a full environmental risk assessment covering potential effects on non-patient populations, biodiversity, and ecosystem exposure pathways. The contained use framework under 2009/41/EC is comparatively structured around a proactive risk classification, with shorter and more predictable assessment timelines.
Understanding which directive governs your submission in each member state is not a detail to confirm late in the process. It determines the technical file structure, the ERA scope, and the competent authority you are dealing with.
The Competent Authority Landscape Is Fragmented — by Design
Adding another layer: the national authority responsible for GMO review is not always the same body that handles the clinical trial authorisation — and it is not always the same authority for contained use versus deliberate release within the same country.
In some member states, the medicines agency holds responsibility. In others, environmental ministries, biosafety committees, or agriculture-adjacent bodies take the lead. In several countries, both a medicines authority and a separate environmental authority are involved, each with its own procedural requirements and timelines.
A few concrete examples:
• Germany: the Paul-Ehrlich-Institut (PEI) handles both CTA and GMO submissions. The PEI has published specific guidance on supporting CTA applications involving GMOs. Paul-Ehrlich-Institut - Clinical Trial Authorisations with genetically modified organisms (GMO) - Paul-Ehrlich-Institut
• France: The Manufacturer of the medicinal product should submit the dossier to the Ministry of Research (Ministère de l’Enseignement supérieur et de la Recherche - MESR) for the agreement/authorization of the manufacturing site. DUO
• Sweden: The Swedish Medical Products Agency (Swedish MPA) and the Swedish Work Environment Authority cooperate regarding the regulation of clinical trials of medicinal products containing or consisting of genetically modified organisms (GMO’s). Medicinal products containing GMO | Biological medicinal products | Manufacturing authorisation | Swedish Medical Products Agency
Knowing which authority governs which framework — in each country where you are running sites — is foundational to building a realistic submission timeline.
The Timeline Problem Is Real — and Country-Specific
GMO application review timelines range from zero to up to 12 months, depending on the country and classification of the GTMP-GMO:
For a multi-country trial, misaligning GMO timelines with CTA timelines can bottleneck an entire programme. And — critically — compliance with the GMO framework must be in place before the clinical trial can start, regardless of where you are in the CTA process.
CTR 536/2014 Didn't Solve It
The Clinical Trials Regulation was a meaningful step forward for harmonizing clinical trial applications. But Article 91 explicitly excludes GMO environmental aspects from CTR scope. A separate GMO application is still required in parallel.
Further practical complications remain:
• Biosafety dossiers cannot be submitted via CTIS — they must go through to national competent authorities.
• While CTR 536/2014 removed GMO authorisation as a formal prerequisite for CTA validation, practical dependencies remain in several countries.
Clinical Site Readiness Is a GMO Variable Too
An often-overlooked dimension of the GMO approval process is clinical site readiness — and it can materially affect both timelines and risk.
Some national frameworks require that individual clinical sites demonstrate specific biosafety capabilities before a GMO IMP can be administered there. This includes appropriate facilities for storage, handling, administration, and disposal of the investigational product, as well as — in some cases — documented shedding management protocols.
Critically, whether a site has previously been approved for a similar GMO product matters. Sites with prior GMO IMP experience and existing national authority recognition can move significantly faster through site-level GMO assessment. Sites without that history may face longer evaluation periods or require additional documentation to demonstrate containment capability.
This makes site selection a regulatory strategy decision, not just an operational one. Choosing sites with existing GMO approval infrastructure — where country frameworks permit — can compress your overall programme timeline meaningfully.
What the Environmental Risk Assessment Actually Requires
The ERA must consider potential adverse effects on:
• Clinical staff involved in administering or handling the product
• Family members and members of the public in direct contact with treated patients
• Animals, plants, microorganisms, and the broader environment
A particular area of focus is shedding — the dissemination of vectors through secretions or excreta. Shedding studies should ideally be integrated into biodistribution or other non-clinical studies early. The data informs the likelihood and extent of human shedding and shapes the environmental transmission risk conclusion.
For viral vector-based GTMPs, additional ERA considerations include replication competence (RCV assays), infectivity, virulence, pathogenicity, and genetic sequence characterisation.
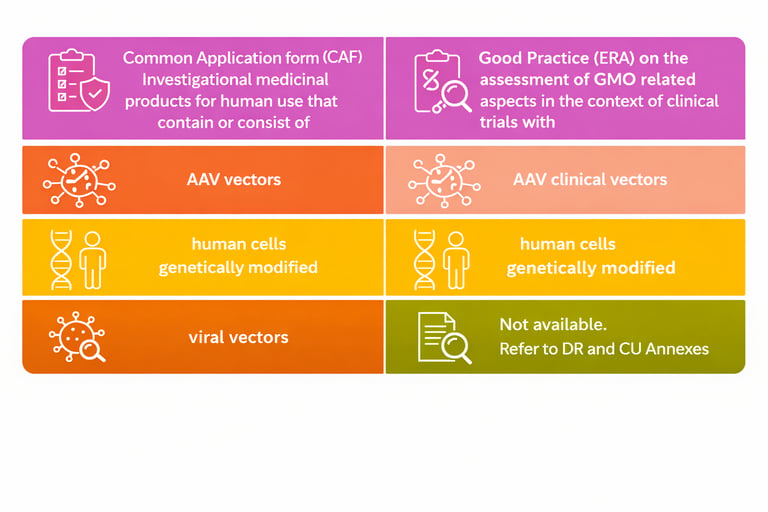
What Is Actually Improving
The EMA ATMP‑GMO Action Plan has established Common Application Forms (CAFs) for three product categories — AAV vectors, other viral vectors, and genetically modified human cells — now accepted across most EU member states. These CAFs, together with the Good Practice (ERA) guidance on the assessment of GMO‑related aspects in clinical trials, form the foundation of the complete GMO dossier.
Some national authorities have also introduced flexible approaches, such as covering multiple clinical trial phases under a single GMO application or applying streamlined procedures for several trials using the same product. These opportunities should be explored proactively to optimise regulatory efficiency and harmonisation across jurisdictions.
The Case for Expert-Led GMO Strategy
Given the complexity above — fragmented competent authorities, country-specific directive selection, variable ERA scope, site-level readiness requirements, and divergent timelines — the value of early expert input is not rhetorical.
An experienced GMO regulatory strategist brings:
• Jurisdiction mapping: knowing which authority governs CU versus DR in each target country, and what their current procedural expectations are
• Dossier calibration: structuring technical files to the correct directive from the outset, avoiding rework that costs months
• Site assessment: Identify which proposed clinical sites already conduct GMO clinical trials and which require additional qualification steps.
• Timeline compression: sequencing submissions to run in parallel where possible, and identifying countries where streamlined or consolidated applications are accepted
• Risk anticipation: flagging country-specific interdependencies before they become programme-level delays
The difference between a reactive GMO strategy and a proactive one is not marginal. For programmes targeting multiple EU countries, it can mean the difference between a 3-month bottleneck and a 12-month one — with direct consequences for patients waiting on access to these therapies.
Strategic Recommendations for Regulatory Affairs Teams
1. Build your GMO roadmap from day one — not after IND-enabling studies are complete
2. Determine which directive applies in each country — contained use, deliberate release, or both — before structuring any technical file
3. Map the competent authority landscape — identify who governs GMO review separately from the CTA authority in each member state
4. Assess clinical site GMO history early — prior site approvals for similar products can significantly compress timelines
5. Integrate shedding studies into non-clinical planning — biodistribution studies with shedding endpoints reduce later standalone study requirements
6. Explore consolidated GMO applications — some authorities accept a single application across phases or sites
7. Engage GMO regulatory expertise early — the framework is complex enough that specialist input at strategy stage pays for itself many times over
The Bottom Line
The EU GMO framework was built for agriculture. Its application to advanced gene therapies remains fragmented, country-dependent, and disconnected from the clinical trial authorisation process.
But it is navigable — with the right expertise, the right sequencing, and a strategy built around the specific directive, authority, and site landscape of your programme.
The patients waiting for these therapies cannot afford for the GMO strategy to be an afterthought.
What has been your experience navigating country-specific GMO frameworks for ATMP clinical trials? I'd welcome perspectives from others working in this space.
Annex: National GMO Requirements by Country
The European Commission maintains a repository of country-specific fiches outlining national regulatory requirements for medicinal products containing GMOs, covering all EU member states, Norway, and the UK. Note: this information has no legal value; relevant national competent authorities should be contacted directly for current requirements.
Genetically Modified Organism (GMO) aspects for investigational medicinal products - Public Health